Since fast and cheap DNA sequencing methods have become firmly established in a wide range of biology the identification of functional elements and transcription factor binding sites (TFBSs) remains an immense challenge.
Because of the limitations of experimental approaches computational bioinformatics are commonly achieved to deal with this analysis demand. This paper uses bioinformatical and statistical analyses to predict TFBSs in DNA sequences [6].
Analysis of Artificial Promoter Constructs
Introduction
The general basis of the varied analyses are 21 short DNA sequences by Katja where, however, different motifs of interest have been placed in. Regarding to a meta-analysis of DNA array data of rodent tissues [2] these motifs are assumed to be binding sites for circadian clock related transcription factors (TFs). The circadian clock controls most of the physiological and behavioural networks by periodic binding of specific TFs to sets of clock genes; thus, the TFs influence biochemical pathways by administering transcription regulation [1].
TFs are sequence specific DNA binding proteins which are crucial trans-acting with cis-regulatory sequences. This interplay is the most important instance of transcription initiation, consequently, common mature bioinformatic approaches address this key moment by identifying TF binding motifs [25,4,22,13]. One characteristic of TF binding is the range of variability of regulatory sites, hence, not all positions of the motifs are strictlyThe general basis of the varied analyses are 21 short DNA sequences by Katja where, however, different motifs of interest have been placed in. Regarding to a meta-analysis of DNA array data of rodent tissues [2] these motifs are assumed to be binding sites for circadian clock related transcription factors (TFs). The circadian clock controls most of the physiological and behavioural networks by periodic binding of specific TFs to sets of clock genes; thus, the TFs influence biochemical pathways by administering transcription regulation [1].
TFs are sequence specific DNA binding proteins which are crucial trans-acting with cis-regulatory sequences. This interplay is the most important instance of transcription initiation, consequently, common mature bioinformatic approaches address this key moment by identifying TF binding motifs [25,4,22,13]. One characteristic of TF binding is the range of variability of regulatory sites, hence, not all positions of the motifs are strictly conserved. Generally, this makes sense biologically that transcription is variable regulated, whereas restriction sites, for example, are not.
Dinucleotide Statistics
A dinucleotide statistic has been chosen for the first characterisation because it is solely statistically motivated and does not assume any underlying biological knowledge about TFBS.
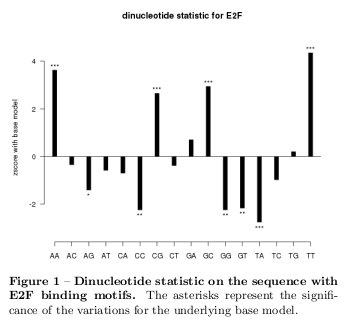
It also suites the type of used sequences because a motif of interest has been placed repeatedly within the random sequence.
The aim of this analysis is to measure the over- and under-representation of two letter words. In contrast to a random sequence there are significant variations expected. In particular, a determining bias in GC content is observed for conserved binding motifs [24] because of CpG suppression: Normally the cytosine of CpG dinucleotides tend to be methylated in vertebrates which leads to a spontaneous desamination of the cytosine to a thymine. As a result, there will be a T-G mismatch which sometimes is repaired to a T=A. On a evolutionary time period this causes underrepresented CpG dinucleotides in non coding DNA.
To measure the dinucleotide representation the nonparametric \( \rho \) statistic (1) is used as a basis for a standardised z-score statistic (2).
\[ \rho_{xy} = \frac{f_{xy}}{f_x \times f_y} \] \[ z_\mbox{score} = \frac{\rho_{xy} - E(\rho_{xy})} {\sqrt{Var(\rho_{xy})}} \]
where \( f_{xy} \) and \( f_{x\vee{}y} \) are the corresponding frequencies to the studied sequence, \( E(\rho_{xy}) \) is the expected mean, and \( Var(\rho_{xy}) \) is the variance according to the given model for the sequence.
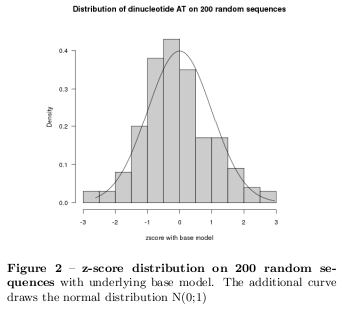
The calculation has been done in Gnu-R using the seqinR package [5] with the base model. Figure 2 shows a \( z_\mbox{score} \) distribution on 200 random sequences and the corresponding normal distribution. With this underlying model z-scores have been calculated on the given sequences. For instance, Figure 1 shows the results for the E2F-sequence with great variations in various dinucleotides as CpG, GcP, TpT, ApA, TpA, and further ones. Clearly, the type of the sequences causes this deviation from the equipartition. The results of the remaining sequences are in the supplementary material.
Finally, overrepresented motifs have been extracted from the sequences if they are longer than 5 bases. This respects the length of the placed motifs and most TFBS in general. For the discussed E2F-sequence the following motif has been predicted:
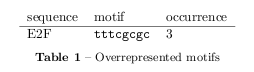
at which is in a perfect accord with the E2F consensus sequence TTNNCG. The remaining results are presented in the supplementary material and it became evidence that most of the correctly found motifs are from highly conserved TFs and this method based on two letter word distributions often fails due to the variation of less conserved TFBSs.
Random Sequences as a Background Model
The dinucleotide statistics used a simple normal distribution as a background model for DNA sequences but by the type of the sequences and the results of the dinucleotide statistic it has been assumed that the statistically independence is a inadequate approximation for a simulated background. For further statistics a better context dependent background model which suits the biological background of the sequences needs to be established.
Mutual Information
To measure the divergence of the sequences from a statistically independent random sequence the mutual information equation [18] has been used: \[ I = \sum_{i;j=1}^\lambda p_{ij} \log_2\frac{p_{ij}}{p_i\times p_j} \] The Taylor expansion the equation leads to a \( \chi^2 \) distributed variable: \[ \hat{I} \approx \frac{1}{2\log_e2} \sum_{i;j=1}^\lambda \frac{(p_{ij}-p_ip_j)^2}{p_ip_j} \] According to John Hawks Web-Blog [10] the significance of mutual information is tested with a Pearson's \( \chi^2 \) test and 9 degrees of freedom: There are 3 independent bases which leads to 9 independent dinucleotides. In a statistically independent random sequence a base provides no mutual information about other bases which leads to a mutual information content of \( I = 0 \). This is however an unrealistic claim for empirical random sequences and the equation already shows a statistically distributed variable. Therefore, a set of 1000 shuffled sequences has been created to calculate a empirical distribution of the test variable and compare this with the theoretical \( \chi^2 \) distribution. The normalization of the empirical distribution is given by: \[ 2n \log_e 2 \] where \( n \) is the number of cases in the sample. [10]
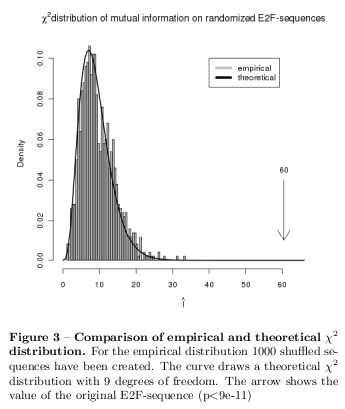
Figure 3 shows the empirical results of the Pearson's \( \chi^2 \) test. As already assumed, the divergence from the statistically independent model is highly significant \( (p<9e-11)\). The remaining results are presented in the supplementary material.
Markov Chain
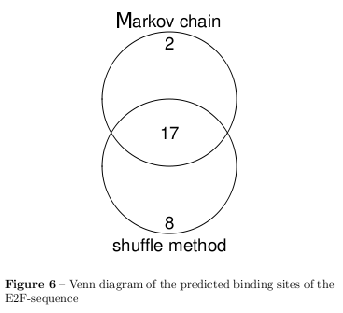
Obviously, the results show that a simple shuffle method which assumes statistically independence would distort the statistically characteristics of the two letter words distribution in the simulated background such as the overrepresented GC content of the E2F-sequence. For this reason, a Markov chain of first order with a one step transition matrix has been used to simulate a background with the same two letter words statistic as the foreground. [8] A markov chain of first order uses initial probability distribution and a one step transition matrix to derive the probabilities for the following bases based on the further one. For representing two letter words a markov chain of first order is needed.
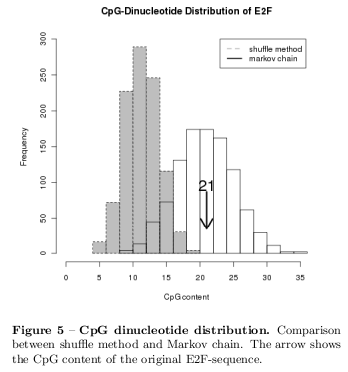
The initial probability distribution \( (\pi^{(0)}) \) of the E2F-sequence is given by: \[ \begin{aligned} \pi^{(0)} &= P(x(p=0)=i) \ \mbox{with } i \in \lambda(a,c,g,t) \\ &= \frac{n_i}{\sum_i^\lambda n_i} \\ \pi^{(0)}_\mbox{E2F} &= (0.1859;\; 0.2756;\; 0.2756;\; 0.2628) \end{aligned} \] The transition matrix is given by: \[ \begin{aligned} p_{ij}(p) &= P(x(p+1) = j\, |\, x(p) = i) \ \mbox{with} i\wedge j \in \lambda \\ p_{ij}(p) &= P = \begin{pmatrix} p_{aa}(p) &\ldots &p_{at}(p) \\ \vdots &\ddots & \vdots \\ p_{ta}(p) &\ldots &p_{tt}(p) \\ \end{pmatrix} \\ P_\mbox{E2F} &= \begin{pmatrix} & a & c & g & t \\ a &0.0774& 0.0452& 0.0258& 0.0387 \\ c &0.0387& 0.0387& 0.1355& 0.0645 \\ g &0.0645& 0.1419& 0.0387& 0.0258 \\ t &0.0000& 0.0516& 0.0774& 0.1355 \\ \end{pmatrix} \end{aligned} \] The probability distribution at position \( (p+1) \) is given by: \[ \pi^{(p+1)} = \pi^{(p)}\times P \] Illustrative, figure 4 shows the process of random sequence generation with a first order Markov process and finally 1000 random sequences have been simulated and as an example figure 5 shows the results of shuffle model and Markov chain simulation in comparison to the CpG content of the original E2F-sequence. The remaining result are presented in the supplementary material
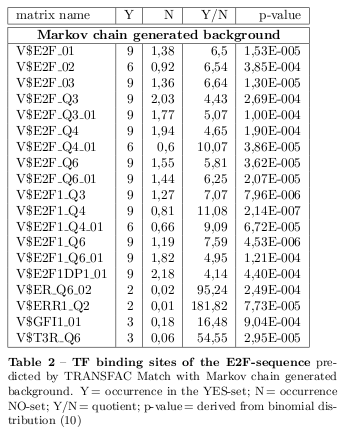
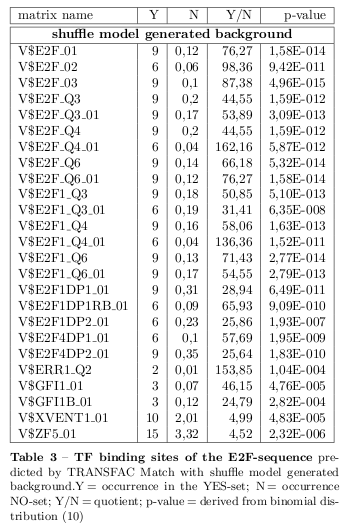
TF Binding Site Search Using Transfac (Match)
In order to find TFs binding potential motifs in the sequences a representation of the binding behaviour of a TF is needed and the possibility to score the binding quality. Therefor, the 21 sequences have been uploaded as YES-set to the analysis system Explain 2.6 of the TRANSFAC suite for a TF binding site search using the Match tool. Additionaly, 1000 randomized sequences for each of the 21 sequences have been provided as NO-set. The sites of the TRANSFAC database are represented by position weight matrices (PWM). A weight matrix derives from a simple position frequency matrix which contains the number of observed bases at each position of aligned binding sites. By converting the normalised frequencies into a log-scale
PWM conversion: \( p(b,i)= \) probability of base b in position i; \( p(b)= \) background probability of base \( b \) \[ W_{b,i} = \log_2\frac{p(b,i)}{p(b)} \]
a motif can be easily scored by summing the position specific values of the weight matrix. The great advantage of PWMs is that summing log-scores is much more handy than multiplying small probabilities. [25] The Transfac-Match program is a matrix-based site prediction thus scores all possible binding sites on the sequence. The threshold of accepted binding scores has been \( \geq 0.73 \) of the maximal motif score and the set of matrices have been the vertebrate: all[minSUM] which minimise the sum of false positive and false negative predictions.
Concluding, the matrices with a p-value \( < 0.001 \) derived from the binomial distribution
Binomial distribution: \( k= \) the number of motifs in the YES-set; \( p= \) motif probability derived from the NO-set; \( n= \) number of potential motif-positions \[ P(k) = \binom{n}{k}\,p^k(1-p)^{n-k} \]
have been selected as final predictions and are presented for the E2F-sequence in tables 2 and 3.
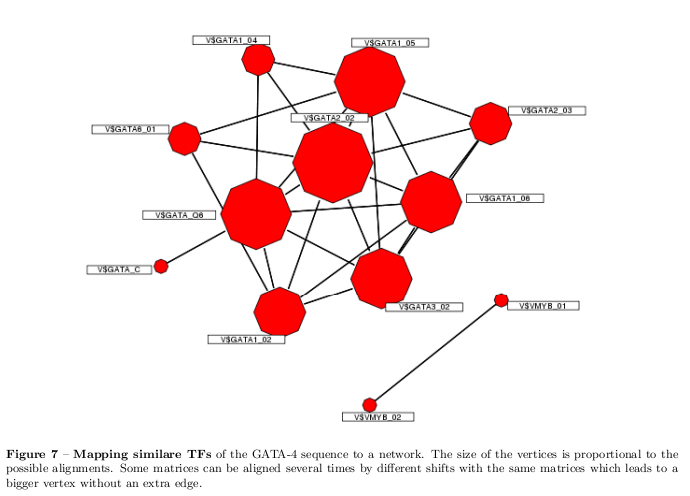
Similarities Between Predicted TFs
To identify highly similar TF binding profiles a \( \chi^2 \) distance and a correlation coefficient for each possible overlap of the PFMs of the predicted TFs have been used. [12] First, has been a set of all possible combinations of PFMs and their iterative overlapping generated. Second, for the entire set have been \( \chi^2 \) distances calculated. The \( \chi^2 \) distance considers the small sample-size of most of the available Transfac PFMs. If the rows are assumed to be statistical independent, the goodness of the alignment can be evaluated with a \( \chi^2 \) distribution with 3 degrees of freedom (because only the occurrence of three bases are independent). The threshold of the alignment used is \( \chi^2_{th}(p=0.05)=7.81 \). If the \( \chi^2 \) distance given by: First, has been a set of all possible combinations of PFMs and their iterative overlapping generated. Second, for the entire set have been \( \chi^2 \) distances calculated. The \( \chi^2 \) distance considers the small sample-size of most of the available Transfac PFMs. If the rows are assumed to be statistical independent, the goodness of the alignment can be evaluated with a \( \chi^2 \) distribution with 3 degrees of freedom\footnote{because only the occurrence of three bases are independent}. The threshold of the alignment used is \( \chi^2_{th}(p=0.05)=7.81 \). If the \( \chi^2 \) distance given by: \[ \chi^2 = \sum_\lambda \frac{N_{g,i}f_{\lambda,i}- N_{f,i}g_{\lambda,i}} {N_{f,i}N_{g,i}(f_{\lambda,i}+g_{\lambda,i})} \ \mbox{with } \lambda=\text{a,c,g,t} \] where \( f \) and \( g \) are the frequencies of the two PFMs, \( i \) is the aligned position of the matrices, and \( N \) is the sample size at the aligned position exceeds the threshold, the aligned position is defined as dissimilar. To simplify the measuring, as done in [12], simply the dissimilar positions are counted. Third, a random sequence is scored with the set of PFMs to calculate correlation coefficients which were compared with the results of the \( \chi^2 \) distance results. For the correlation coefficient the experimentally verified binding motifs of the PFMs are used to construct PWMs, where the weights are calculated as: \[ \begin{aligned} p_{\lambda,i} &= log_2\frac{f_{\lambda,i} + \epsilon_\lambda}{N_i, + \sum_\lambda \epsilon_\lambda} \\ w_{\lambda,i} &= \frac{p_{\lambda,i}}{r_\lambda} \end{aligned} \] where \( \epsilon_\lambda \) is a pseudocount \( \epsilon_\lambda=\frac{\sqrt{N_i}}{4} \) and \( r_\lambda \) refers to the expected a priori probability. The used pseudocount is motivated by the coefficient of variation. The score of a motif is represented by the sum of the weights over all positions \( i \). The Pearson correlation coefficient is given by: \[ PCC(f,g) = \frac{\sum_i^n (f_i-\bar{f})(g_i -\bar{g})}{(n-1)s_fs_g} \] where \( f \) and \( g \) are the scores at the position \( i \), \( \bar{f} \) is the sample mean, and \( s \) is the sample standard deviation. Therefore, the PCC gives a measure of agreement between the two sets of scored sequence by their means of covariance. All aligned TFs with less than two dissimilar positions and a correlation coefficient greater than 0.8 are arranged in a network as shown in figure 7.
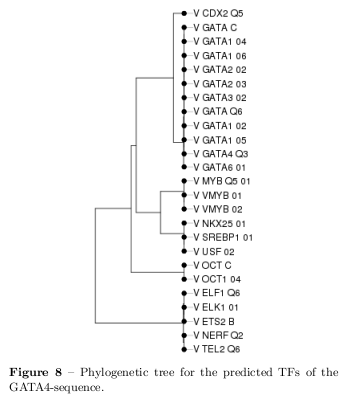
Phylogenetic Tree Construction
The STAMP web interface [15] has been used for an ungapped multiple alignment of the predicted PFMs and a phylogenetic tree has been constructed [9]. As similarity metric the Pearson correlation coefficient, and for the multiple alignment the ungapped Smith-Waterman algorithm for local alignment has been used. In reference to [14], this combination reaches the best performance at a evaluation of similarity metrics and motif alignment strategies. The Smith-Waterman algorithm is based on dynamic programming and finds the best scoring local alignment by iterative calculating position scores based on previous scores. Finally, the unweighted pair group method with arithmetic mean (UPGMA) for clustering and phylogenetic tree construction has been used. Thus, the tree is constructed by progressively clustering the most similar TFs at each time step beginning with the input sequences which serve as leaves. The distance matrix is in the PCC metric and the distance of a cluster is calculated by the mean distance between each elements of the compared clusters. \[ d(G_i,G_j) = \frac{1}{N_iN_j} \sum_{x\in G_i} \sum_{y\in G_j} d(x,y) \] where \( N \) is the number of elements in a group \( G \). The UPGMA algorithm can be explained by the following pseudo code:
- at the beginning all TFs of the input serve as leaves
-
while( number of groups \( >1 \) ) {
- merge those \( G_i \) and \( G_j \) with the smallest distance: \( d(G_i,G_j) = \min \)
- the new root of the merged groups has the mean distance between each elements of the groups \( G_i \) and \( G_j \): \[ \frac{d(G_i,G_j)}{2} \]
- for all remaining ellements \( k \): \[ \forall k: \ d\,' (G_k,(G_i,G_j)) \]
It should be noted that the UPGMA algorithm assumes the molecular clock hypothesis which describes a constant rate of change for all leaves in the phylogenetic tree. Hence, all leaves have the same distance to the finale root of the built tree. This means that the initial distance matrix has to be ultrametric: If two random elements have the same distance to a third one, the distance to each other have to be shorter. Otherwise, the resulting tree built by the UPGMA algorithm has a variational distance matrix because it always constructs an ultrametric tree.
\[ \begin{aligned} \forall\{x,y,z\}: & d(x,y) = d(y,z) \\ & \rightarrow d(y,z) < d(x,z) \wedge d(y,z) < d(x,y) \end{aligned} \]

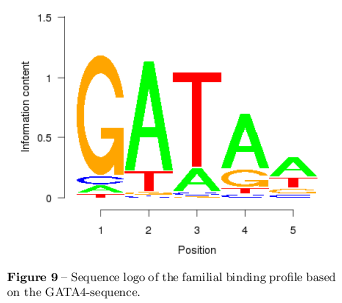
Sequence Logos
Sequence Logos are a great graphical method for displaying aligned sequences or binding profiles and combines different information as the consensus sequence, relative frequencies, the amount of information, and particular locations [20]. To generate a sequence logo, firstly, the sequences are aligned relative to one another, secondly, the frequency of each base at all positions is determined. Because the consensus sequence does not provide the complete information about sequences it is a poor model for representing binding sites. Therefore, the information content of a binding site position is a more efficient approach which describes the average required dichotomous questions needed to be answered on a optimal strategy. This is given by the Shannon Entropy: \[ H_1 = \sum_i^\lambda -p_i \log_2p_i \] where \( p \) is the probability and per definition: \[ \begin{aligned} \lim_{p\rightarrow 0} p \log_2p = & \lim_{p\rightarrow 0}\frac{(\log_2p)'}{(-\frac{1}{p})'} = 0 \end{aligned} \] However, it is counterintuitive to use the Shannon Entropy to represent binding profiles because conserved positions have the smallest entropy. Hence, the height of letters in a sequence logo is defined as: \[ R_\mbox{seq} = H_\mbox{max} - H_1 \] where \( H_\mbox{max} \) is the maximal possible entropy which is 2bit for a DNA sequence.
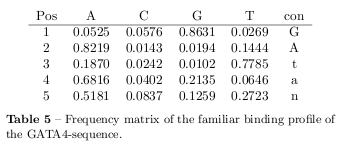
The frequency matrix of the aligned binding profiles predicted by MATCH on the GATA4-sequence is shown in table 5 and the resulting sequence logo is shown in figure 9.
Discussion
The analysed sequences show a great statistical divergence from random sequences which has been shown in several statistical tests as the \( \chi^2 \) test of mutual information. Also the first dinucleotide statistic shows strong deviation from the random model which actually can be used to predict motifs within the sequences. This reason makes it crucial to choose a better background model than a random one. Specially to respect the higher CpG content of motifs a markov model has been chosen to generate a background of 1000 randomised sequences per sequence for the following TFBS prediction. The initial list of TFs binding the sequences has been several hundred TFs long. Bearing in mind that most of the PWMs have a low information content, the list was reduced to TFs with high complexity with the help of the background model. The choice of the background model shows a determining effect on the finally predicted TFs which is shown in figure 6. The final list of predicted TFs is assumed to contain TFs with very similar binding behaviour. By arranging similar TFs to a network it has been shown that the predicted TFs cluster with the motifs in the sequence. (q.v. figure 7) This clusters can be arranged to family binding profiles by a multiple alignment of the individual PWMs which has been shown for the GATA binding motif in figure 9. The results of this analysis are in agreement with the motifs placed into the sequences. The choice of the background model and the used thresholds for rejecting TFs are crucial and have a strong impact on the rate of false-positives and false-negatives. The used values have been confirmed to perform best by many meta studies and are chosen for further analysis.
Analysis of experimental verified circadian sequences from the Schibler laboratory, Switzerland
Hans Reinke et al. [19] used a random DNA library to find circadian TFs on an experimental approach. Probes of liver nuclei of mice were prepared at different Zeitgeber times and used for Electrophoretic Mobility Shift Assay (EMSA) reactions. The assay validates protein-DNA binding because complexes of DNA and bounded proteins are less mobile in the electrophoretic separation than the control lane without proteins. With this approach they found 43 probes which show circadian DNA-binding activity with two different probes and finally sequenced the underlying DNA sequences.
Preparing the Sequences and Characterisation
The library consists of twice 50bp random sequences and a 5'-flanking region with a AscI restriction site and a palindromic 3'-flanking region containing a BamHI restriction site.
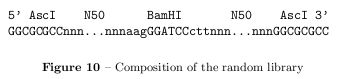
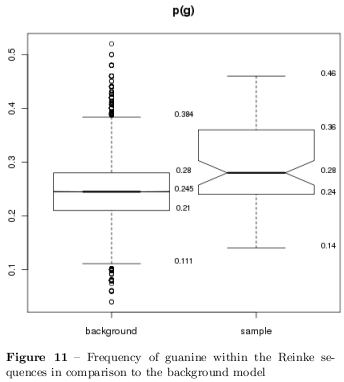
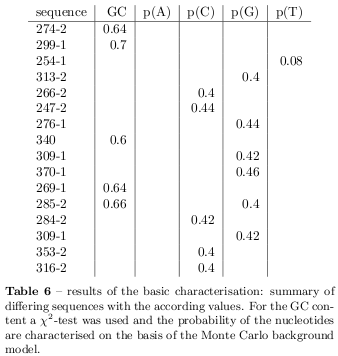
Because there have been no known motifs from the TRANSFAC database been found within the flanking regions, the sequences have been adjust and redundancy removed. To unify the data all the probes have been renamed with the number of the random sequences presented in the Supplemental~Table~1 in the Reinke et al. paper [19], whereat the \( N50 \) at the \( 5' \)-end is called "-1" and the second \( N50 \) sequence close to the \( 3' \)-end is called "-2". With one exception, probe 82 from the Reinke data could not be aligned with the sequences presented in the Supplementary Table 1 and is called UNKNOWN. In case the circadian binding activity could not be narrowed to one of the random sequences the entire plasmid sequence is used and can be recognised at numbers without a specifying suffix. A Monte-Carlo background model with 1000 random sequences of the same length has been used to define the expectation interval of 95% of nucleotide frequency in random sequences and finally significant frequencies have been characterised (with \( p<0.05 \) ). Figure 11 shows the data for the guanine frequency within the Reinke sequences and the background model and it is evidence that the frequencies within the Reinke sequences differ from the background model.
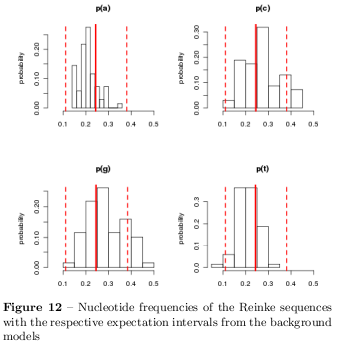
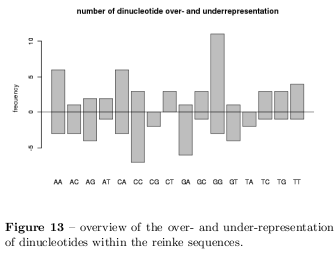
In figure 12 it can be realised that specially the GC content of the sequences should be demonstrative and in order to characterise the GC content a \( \chi^2 \) test was used to distinguish sequences which differ significant with \( p<0.05 \) what is the case for 5 sequences. Table 6 shows the summarised results of the characterisation based on the Monte Carlo background. The sequences differ primarily in their GC content. Surprisingly, not even one of the sequences shows any irregularities in their CpG dinucleotide statistics as introduced before. Figure 13 summarises the results of the dinucleotide statistics where positive frequencies represent the number of sequences with overrepresented dinucleotides and negative frequencies represent under-representation. Specially the GpG dinucleotide is very often overrepresented and CpC dinucleotides are specially underrepresented. To find PolyA stretches a window of different sizes has been slided over the sequences scoring possible stretches whereat the following stretches has been found:
- 254-1: acaaaaa
- 350: tatttgttat
- 335: ttatttatt
- 391: aaactaaa
- 378-1: tttattat
- 391-1: aaactaaa
- 152-2: aataataa
- 398-2: aacaaaa
For choosing a suitable background model, the mutual information content has been calculated as described before. But only 3 sequences exceed the significance criterion of \( p<0.05 \) which are sequence 276-1, 285-2 and 378-1 where a the earlier introduced markov chain was choosen. For all the other sequences a simple identical, independent distributed (iid) background model was used.
TFBS prediction with Transfac
Reinke et al. found experimentally confirmed TFBSs which still showed a circadian behaviour but had some deviation from the motifs consensus sequence. With the help of the PCMs from Transfac the percental deviation of the motifs was calculated.
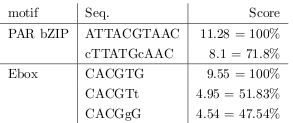
Because the TRANSFAC search earlier has performed best on a threshold of \( \geq 0.73 \) of the maximal matrix score this threshold has been chosen also for this TRANSFAC search. This is also in accordance with the results of the experimental confirmed PAR bZIP motif with two mismatches. Unfortunately this threshold is to strict for the Ebox mismatch motifs but a threshold of 50% would raise to many false positives and is not workable. In regards to a medium strength conserved binding motif of length greater than 8 this threshold reflects approximately 2 allowed mismatches.
Results
As before, all the predicted TFBSs with \( p<0.001 \) have been assembled to final lists of predicted TFBSs which are presented in detail in the supplementary material. All matrices are presented with their consensus score and the information content of the matrices in bit. The information content is reflective of the quality of the matrices and the expected distance of a motif within the background model is given by:
\[
\text{distance} \approx 2^{\mbox{score}}~ \mbox{bp}
\]
So is the TRANSFAC matrix V_CDXA_01 of bad quality and has only a information content of 4.8bit which leads to an expected distance of \( 2^{4.8} \approx 28\,\mbox{bp} \) where instead the matrix V_P53_01 has a much better quality with a information content of 33.78bit which leads to an expected distance within the background model of \( \approx 1.5\mbox{E}10\,\mbox{bp} \).
Table 7 shows the most frequently predicted TFBSs. The binding motif USF_C is very similar to the other USF motifs and correlates also with several other Ebox motifs such as CLOCKBMAL, EBOX and MYCMAX. The USF motifs does not match any known transcription factor but the similarity analysis classifies them as a Ebox motif. In summary most of the most frequently predicted TFBSs are Ebox motifs which is in agreement with the results by Reinke et al.
Sequence 273-1 is predicted as a Clock/Bmal binding motif by Reinke et al. however I found no overrepresented Ebox motif within this sequence. Instead I predicted a PPAR \( \alpha \) (proliferator-activated receptor alpha) motif, a member of the nuclear receptor family of ligand activated TFs, and is a key regulator of lipid metabolism by regulating the peroxisomal beta-oxidation pathway. This motif clusters also with the predicted vitamin D receptor (VDR) which is co-localised with the Sp1 transcription factor gene on human chromosome arm 12q. [23] It has also been shown that PPAR \(\alpha \) is regulated by CLOCK and regulates Bmal1 expression itself. [27]


Also in sequences 297-2 and 353-2 were no overrepresented Ebox motifs predicted but a YY1 motif instead which is the target of the YY1 transcription factor. The ubiquitous transcription factor Yin Yang 1 (YY1) is known to play a fundamental role in several processes and is also known to be associated with cell cycle phases. Recent studies have also shown that YY1 seems to be implicated in the regulation of tumor cell resistance to chemotherapeutics. A circadian activity of this TF can be important in chemotherapy scheduling.
In sequence 270-1 are several AP1 motifs predicted. The activator protein 1 is a growth factor sensor which controls cellular processes as differentiation, proliferation, and apoptosis by binding to the TPA DNA response element. AP1 is one of the best studied target of MAPK signalling whereat the MAPK pathways are found to be connected to the circadian clock. [7]
The cAMP response element-binding protein (CREBP) predicted in sequences 101-1 and 284-2 is also known to be circadian regulated at least in gene expression of suprachiasmatic nuclei which is the central pacemaker of the circadian clock. [17]
The sequences 398-2, 244-1, 246-1 and 316-2 are in accordance with the Reinke et al. results predicted Ebox motifs.
Also the results of the HSF1 could be replicated for sequences 317-1 and 325-2.
The results of sequence 98-1 are very interesting: With HLF and the avian TEF ortholog VBP are truly PAR-bZip motifs predicted as by Reinke et al. But beside of the PAR-bZip motif a CEBP motif is predicted and the best scoring E4BP4 TF has a known antagonistic role in the circadian oscillatory mechanism 16] and does not cluster with any other predicted TF of this sequence.
In Table 8 all the predicted motifs are compiled which are also predicted in the Bozek et al. article [3]. In summary 56% of the predicted motifs could be replicated within the Reinke sequences. Specially several liver enriched TFs with clear circadian mRNA patterns as HNF1, C/EBP \(\alpha \) and - \(\beta \) and Arnt, CLOCK/Bmal and PARbZip motifs have been predicted. [27]

Interesting new results are for example zinc finger proteins HELIOS and IK-2 which cluster together and are predicted together with E2F, which plays a important role in the cell cycle, and SRF. The serum response factor (SRF) participates in the regulation of cell cycle, cell growth, cell differentiation and apoptosis and is the downstream target of several pathways such as MAPK which is found to be clock related. [7] Ikaros binding elements have been found in the 5' flanking region of STAT4 (signal transducer and activator of transcription) and plays an important role in CD4 versus CD8 differentiation.
E2F is a key regulator of cell proliferation, GABP is a major contributor to cell growth, the tumor repressor protein P53 is known for regulating the expression of stress response genes and mediates anti-proliferative processes.
The NKX2.5 is a homeobox-containing TF which functions in heart formation and development and cooperates with GATA4 as a activator of the atrial natriuretic factor gene (ANF). ANF is a peptide hormone and is involved in homeostatic control whereat a study has shown that pro-hormone peptides show circadian rhythm in man. [26]
The ATF4 TF clusters with CREBP and was the only predicted motif in sequence 378-1. ATF4 is a direct target of CLOCK and seems to play a role in drug resistance in human cancer cells. [11]
In sequence 391-1 AT rich motifs has been predicted where YY1 and POU3F2 are similar to CDXA. POU3F2 is a CNS-specific TF.
The predicted TTF-1 TF contains a homeodomain and regulates the circadian rhythm of pituitary adenylate cyclase-activating polypeptide gene expression in the rat hypothalamus and also colocolizes with Per2, a circadian feedback loop controller within the SCN. [21]
Summary
Reinke et al. ascribe the observed circadian activity to Rev-ERB \( \alpha \), PARbZip, ClockBmal and HSF1 whereat the HSF1 is described for the first time to be circadian. But based on different meta-studies it is assumed to find more promising TFs which show circadian behaviour. [2] [3] Using GC matched background models additional TFs are predicted within the sequences. Most of them are known as Ebox, Dbox and Y-box motifs. A great fraction of the TFs from the Bozek et al. papers are also predicted within the Reinke sequences which argues for the corresponding TFs. Specially within some of the sequences which have been characterised as HSF1 by Reinke et al. were well known circadian TFs found as PPAR \(\alpha \) and E4BP4.
But also new promising candidates for circadian TFs were predicted as HELIOS and IK-2, the serum response factor (SRF), E2F, TBP, IPF1, AP1, GATA, HNF-1 and TFs of the POU family as OCT1,4.
Bibliography
- 1
-
ALBRECHT, U.
Regulation of mammalian circadian clock genes.
Journal of Applied Physiology 92 (March 2002), 1348-1355. - 2
-
BOZEK, K., REL´OGIO, A., KIELBASA, S. M., HEINE, M., DAME, C., KRAMER, A., AND HERZEL, H.
Regulation of clock-controlled genes in mammals.
PLoS ONE 4 (March 2009), 1-11.
corresponding author: Herzel, ITB HU-Berlin, h.herzel@biologie.hu-berlin.de. - 3
-
BOZEK, K., ROSAHL, A., GAUB, S., LORENZEN, S., AND HERZEL, H.
Circadian transcription in liver.
Preprint submitted to Elsevier November 23, 2009, November 2009. - 4
-
BULYK, M. L.
Computitional prediction of transcription-factor binding site locations.
Genome Biology 5 (2003), 201.1-201.11. - 5
-
CHARIF, D., AND LOBRY, J.
SeqinR 1.0-2: a contributed package to the R project for statistical computing devoted to biological sequences retrieval and analysis.
In Structural approaches to sequence evolution: Molecules, networks, populations, H. R. U. Bastolla, M. Porto and M. Vendruscolo, Eds., Biological and Medical Physics, Biomedical Engineering. Springer Verlag, New York, 2007, pp. 207-232.
ISBN : 978-3-540-35305-8. - 6
-
COLLINS, F. S., GREEN, E. D., GUTTMACHER, A. E., AND GUYER, M. S.
A vision for the future of genomics research.
nature 422 (April 2003), 835-847. - 7
-
DE PAULA, R. M., LAMB, T. M., BENNETT, L., AND BELL-PEDERSEN, D.
A connection between mapk pathways and circadian clocks.
Cell Cycle 7, 17 (September 2008), 2630-2634. - 8
-
DEONIER, R. C., TAVAR`E, S., AND WATERMAN, M. S.
Computational Genome Analysis.
Springer, 2005. - 9
-
DRAY, S., AND DUFOUR, A.
The ade4 package: implementing the duality diagram for ecologists.
Journal of Statistical Software 22(4) (2007), 1-20. - 10
-
HAWKS, J.
Information theory and mutual information between genetic loci.
Web-Blog, October 2008. - 11
-
IGARASHI, T., IZUMI, H., UCHIUMI, T., NISHIO, K., ARAO, T., TANABE, M., URAMOTO, H., SUGIO, K., YASUMOTO, K., SASAGURI, Y., WANG, K. Y., OTSUJI, Y., AND KOHNO, K.
Clock and atf4 transcription system regulates drug resistance in human cancer cell lines.
Oncogene 26 (2007), 4749-4760. - 12
-
KIELBASA, S. M., GONZE, D., AND HERZEL, H.
Measuring similarities between transcription factor binding sites.
BMC Bioinformatics 6:237 (September 2005), 1-11. - 13
-
MACLSAAC, K. D., AND ERNEST, F.
Pratical strategies for discovering regulatory dna sequence motifs.
PLoS Computational Biology 2 (April 2006), 201-209. - 14
-
MAHONY, S., AURON, P. E., AND BENOS, P. V.
Dna familial binding profiles made easy: Comparison of various motif alignment and clustering strategies.
PLoS Computational Biology 3(3):e61 (March 2007), 0578-0591. - 15
-
MAHONY, S., AND BENOS, P. V.
Stamp: a web tool for exploring dna-binding motif similarities.
Nucleic Acids Research 35 (April 2007), 253-258. - 16
-
MITSUI, S., YAMAGUCHI, S., MATSUO, T., ISHIDA, Y., AND OKAMURA, H.
Antagonistic role of e4bp4 and par proteins in the circadian oscillatory mechanism.
Genes & Development 15 (2001), 995-1106. - 17
-
OBRIETAN, K., IMPEY, S., SMITH, D., ATHOS, J., AND STORM, D. R.
Circadian regulation of camp response element-mediated gene expression in the suprachiasmatic nuclei.
THE JOURNAL OF BIOLOGICAL CHEMISTRY 274, 25 (June 1999), 17748-17766. - 18
-
PŚ
EREZ-CRUZ, F.
Estimation of information theoretic measures for continuous random variables.
In Neural Information Processing Systems (Decemver 2008). - 19
-
REINKE, H., SAINI, C., FLEURY-OLELA, F., DIBNER, C., BENJAMIN, I. J., AND SCHIBLER, U.
Differential display of dna-binding proteins reveals heat-shock factor 1 as a circadian transcription factor.
Genes & Development 22 (2008), 331-345. - 20
-
SCHNEIDER, T. D., AND STEPHENS, R. M.
Sequence logos: A new way to display consensus sequences.
Nucleic Acids Res. 18 (1990), 6097-6100. - 21
-
SON, Y. J., YUN, C. H., KIM, J. G., PARK, J. W., KIM, J. H., KANG, S. G., AND LEE, B. J.
Expression and role of ttf-1 in the rat suprachiasmatic nucleus.
Biochem. Biophys. Res. Commun. 380 (2009), 559-563. - 22
-
STORMO, G. D.
Dna binding sites: representation and discovery.
Bioinformatics 16, 1 (2000), 16-23. - 23
-
SZPIRER, J., SZPIRER, C., RIVIERE, M., LEVAN, G., MARYNEN, P., CASSIMAN, J.-J., WIESE, R., AND DELUCA, H. F.
The sp1 transcription factor gene (sp1) and the 1,25-dihydroxyvitamin d3 receptor gene (vdr) are colocalized on human chromosome arm 12q and rat chromosome 7.
Genomics 11, 1 (1991), 168 - 173. - 24
-
TABACH, Y., BROSH, R., BUGANIM, Y., REINER, A., ZUK, O., YITZHAKY, A., KOUDRITSKY, M., ROTTER, V., AND DOMANY, E.
Wide-scale analysis of human functional transcription factor binding reveals a strong bias towards the transcription start site.
PLoS one 8 (August 2007), 1-14. - 25
-
WASSERMAN, W. W., AND SANDELIN, A.
Applied bioinformatics for the identification of regulatory elements.
Nature 5 (April 2004), 276-287. - 26
-
WINTERS, C. J., SALLMAN, A. L., AND VESELY, D. L.
Circadian rhythm of prohormone atrial natriuretic peptides 1-30, 31-67 and 99-126 in man.
Chronobiology International 5, 4 (1988), 403-409. - 27
-
ZHANG, Y.-K. J., YEAGER, R. L., AND KLAASSEN, C. D.
Circadian expression profiles of drug-processing genes and transcription factors in mouse liver.
DRUG METABOLISM AND DISPOSITION 37, 1 (2009), 106-115.