An analysis of the structural context of microRNA seed sites within 3’UTR sequences points to certain microRNA families which show enriched seed sites within hairpin structures. A sub-set of these hairpins are enriched by AU rich PUM2 binding motifs.
PUM2 seems to be a specific microRNA/hairpin targeting RNA binding protein. The scoring of all 3’UTR sequences supports that PUM2 binding sites are over-represented within hairpins which include microRNA seeds. The gene ontology of genes incorporating microRNA seeds within hairpins yields up an interrelation to the PcG protein complex and the regulation of mesenchymal cell proliferation.
Introduction
The classical dogma of Molecular Biology described a directed one-way relationship of gene regulation through Genes \( \rightarrow \) Transcriptome \( \rightarrow \) Proteome. Gene regulation turned out to be much more complex and diverse with the understanding of gene regulatory networks. Thus networks enable alternative paths as DNA interacts indirectly through its RNA or protein expression products with other DNA segments. The dogma therefore needed to be expanded.
Regulation of gene expression can act on several stages of the DNA-protein pathway: Regulation of Transcription, RNA Processing, RNA Transport and Translation. The majority of genes are protein coding and are controlled at first stage by regulation of transcription rates either through chromatin arrangement and histone modification or through the activity of transcription factors.
Transcription initiation in eukaryotes requires the interaction of transcription factors forming the transcription initiation complex [Kadonaga1998]. This initiation can be regulated by specificity factors such as the sigma factors [Moran1989] or repressors impending the RNA Polymerase progress. A group of activators increases the promoter binding attractiveness and enhancers initiates secondary structure changes in order to bring the promoter into reach of the initiation complex [Serfling1985].
At second stage, genes are controlled at RNA level by post-transcriptional regulation. This includes alternative splicing, degradation by the exosome complex, processing (capping, splicing, polyadenylation and RNA editing), nuclear export and translation. Post-transcriptional regulation gained more attention lately as it plays a larger role than previously assumed [Mata2005]. It has been shown that the amount of processed RNA within the nucleus and cytoplasm differ greatly which gave raise to the magnitude of post-transcriptional regulation [Schwanekampa2006]. The main regulatory processes probably are RNA binding proteins (RBP) and RNA interference (RNAi). RNAi is an RNA-dependent gene-silencing pathway and the two possible RNAs interacting are siRNAs and microRNAs and with increasing 1733 human mature microRNAs reported in miRBase 17, RNAi became a major regulatory pathway. The RNA-induced silencing complex (RISC) has a catalytic component called Argonaute protein which loads the siRNA or miRNA and cleaves the target mRNA. There is also a possible interaction with DNA called RNA-induced transcriptional silencing (RITS). The RNAi pathway exists in metazoa as well as in plants. The main difference lays within the microRNA biogenesis: in the case of plants the microRNA biogenesis is located entirely within the nucleus and is processed by a single enzyme called Dicer-like 1. The final duplex is then exported into the cytoplasm where the mature microRNA is incorporated into RISC.
microRNA biogenesis in metazoa in the context of cellular compartmentalisation. The biogenesis involves several steps which are catalysed by Drosha and Dicer. The product is a duplex where the mature miRNA gets incorporated into the RISC complex. The targeted mRNA gets either translationally repressed or degraded. (Attribution: Narayanese)
Messenger RNAs (mRNAs) are linking the genetic information of the DNA with the synthesis of proteins as an interstage product between transcription and translation. Within the information flow from DNA to proteins, the coding strand of DNA gets transcribed to mRNA. RNA is along with DNA and proteins one of the three major macro molecules essential to all forms of known life. As DNA, RNA is composed of nucleotides in which are four different types: adenine (A), cytosine (C), uracil (U) and guanine (G). Each nucleotide is bound in 3' direction through its phosphate group with the ribose of the next, forming a single stranded chain.
As a necessity of rapidly changing conditions all steps of gene expression are strictly controlled and targeted by many regulatory processes. In case of mRNAs, one post-transcriptional regulator are microRNAs. Micro (mi)RNAs are small RNA molecules typically of size of 22 nucleotides. They derive from primary miRNA gene transcripts called Pri-miRNA which get processed by Drosha into Pre-miRNA. The Pre-miRNAs are exported into the cytoplasm where they get processed by Dicer into miRNA:miRNA* duplexes. After unwinding the dsRNA duplex, the mature miRNA is available. Bound to Argonaute proteins forming the RISC assembly, miRNAs interact with the 3'-untranslated region (3'UTR) of mRNAs and regulate these through two main mechanisms: mRNA degradation and mRNA translation inhibition. The interaction is initiated through the 3'UTR binding site forming hybrids with the miRNA seed region (positions 2 to 8 in the miRNA) [Eulalio20089]. In a similar manner, RNA-binding proteins (RBPs) interact with 3'UTRs as regulators through a sequence specific binding and can both stimulate and inhibit gene expression [Kedde2008].
Even predicted miRNA targets highly correlate with gene expression suppression [Lim2005] not all expression changes are explained by predicted miRNA targeting. It has been shown that RBPs are also correlated with post-transcriptional regulation and are globally coupled to effective microRNA target sites [Jacobsen2010].
It is evident that RBPs play a crucial role within post-transcriptional regulation and mRNAs interact with hundreds of them [Anantharaman2002]. Also, correlation analysis showed that RBPs need to be considered in microRNA target prediction and interpretation [Jacobsen2010]. A question still open is on which level these RBPs and certain microRNAs interact and what the underlying mechanisms are. Main focus laid on site accessibility in microRNA target prediction which improved the prediction of binding sites [Long2007,Rabani2011]. A common used model is based on the free energy gained from formation of microRNA-target duplex and the energetic cost of unpairing the target for accessibility ( \( \Delta\Delta{}G \) ) [Kertesz2007]. In Kedde2010 a novel mechanism of a RBP-induced structural switch mediating microRNA expression was introduced, combining the common understanding of target site accessibility and target site structure. Kedde et al. showed that miR-221/222 are not functioning at their full capacity towards CDKN1B 3'UTR as a result of target site obstacle. Using micro-injection of short RNAs containing both 3' and 5' fluorophores they were able to show that Pumilio-1 (PUM1) induces a local structural change of the target site in a FRET experiment. So far, it is the only example of an RBP modulated structural switch in mRNA. As concluded by Kedde et al., more examples are expected due to the generally high conservation of 3'UTR regions. The aim of this thesis will be to predict further potential candidates for an interaction of RBPs with the structural composition of microRNA target sites within 3'UTR sequences.
Material 3'UTR Sequences and miRNA seed sites
For the further analysis, 3'UTR sequences from the TargetScan Human 5.2 database were used. In case a gene has multiple transcripts, the transcript with the longest 3'UTR was selected for the Targetscan database. The set consists of 17840 3'UTR sequences in total. The database of predicted miRNA target sites was also downloaded from the TargetScan Human 5.2 database. Though, only the set of conserved and broadly conserved miRNA families was considered. Whereby conserved families are conserved across most mammals up to placenta mammals and broadly conserved families include miRNAs conserved across most vertebrates including zebrafish [Friedman2009]. Additionally, only conserved miRNA target sites are incorporated into the database. Site conservation, as used by Targetscan, is defined by conserved branch length. This score is the sum of phylogenetic branch length of species containing target sites as described by Friedman et al. This score is corrected for the individual 3'UTR conservation. The downloaded set contains 54979 predicted microRNA target seed sites.
Correlation between miR seed site accessibility and over-represented words
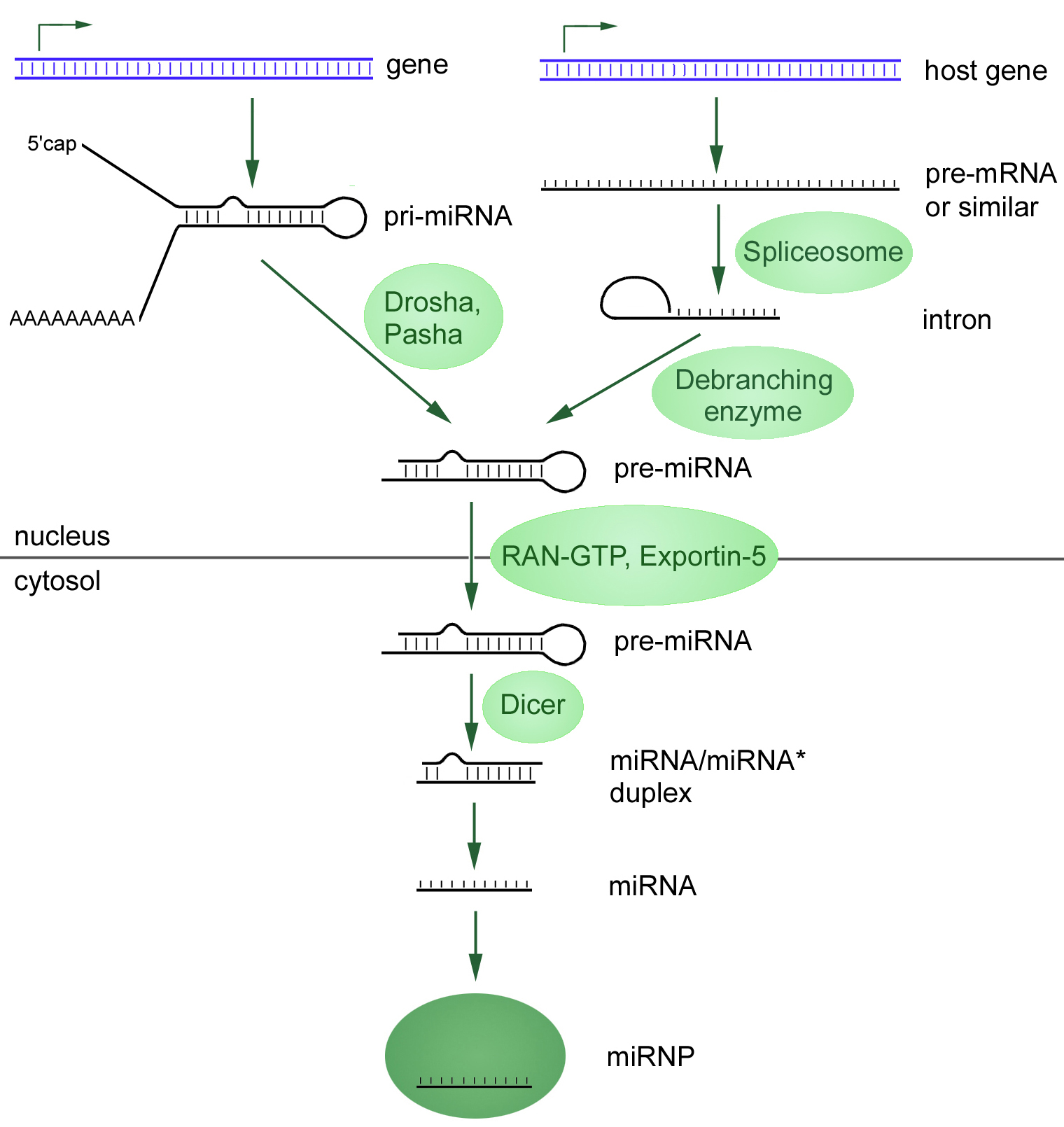
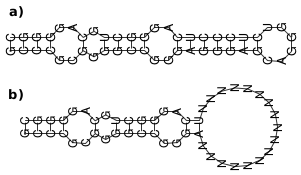
A first approach was to look for correlation between microRNA seed site accessibility and over-represented words. For each of the 54979 predicted microRNA seed sites the corresponding mRNA sequences were extracted containing flanking regions of plus/minus 70nt. This led to a set of 51323 sequences of length 147nt. The discrepancy is due to sites that are too close to the UTR ends to extract full-length flanking regions. As target sites are very dense the extracted sequences may overlap. Because the used method for discovering over-represented words is based on word occurrence in a sorted list, this overlap will bias the statistic of word representation due to artificial enrichment. To reduce the redundancy all extracted sequences are represented as a graph where nodes are connected by edges if the sequences start coordinates are closer than 100nt. The maximal independent set of this graph are vertices which do not have a common edge and are not a subset of any other independent set. The implementation was done using NetworkX 1.5 and led to a set of 15778 sequences.
Maximal independent set of microRNA target sites: Extracting the set of seed sites with flanking regions of 70nt leading to sequences of 147nt length. This sequences can overlap and introduce redundancy. A graph represents all the extracted sequences where edges are drawn if sequences are closer than 100nt. Graph theory is used to extract the maximal independent set which is [1,3,4] in this example.
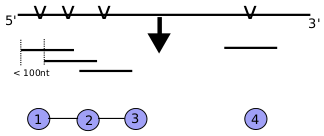
The sequences were ranked by their global accessibility score. This score tries to reflect the microRNA target site accessibility which is important for binding specificity. The score is derived using the RNAfold 1.8.5 software package and is described in Leibovich2010. The global accessibility score ( \( GA \) ) of sequence \( S_i \) is calculated as \[ GA(S_i) = \Delta G_{open}(S_i) + \Delta G_{all}(S_i) \] where \( \Delta G_{all} \) is the minimum free energy (mfe) of the 147nt long sequence \( S_i \). \( \Delta G_{open} \) describes the free energy loss by opening the structure and is defined as: \[ \Delta G_{open}(S_i) = \Delta G_{all}(S_i) - \Delta G_{masked}(S_i) \] To calculate this free energy loss it is necessary to force the target site into an opened structure by masking 25nt by "N" with the seed site centred. The free energy of this masked sequence is called \( \Delta G_{masked}(S_i) \). The motivation to use ranked lists by the global accessibility is that functional seed sites in highly inaccessible regions probably require a cooperative activity of RNA binding factors. Their binding motifs will be imbalanced within the ranked lists due to their correlation with the accessibility score.
Forcing a RNA fold a) into an open structure b) by masking nucleotides by "N".
Distribution of \( GA(S_i) \) of the maximal independent set of 15778 target sites.
Cwords is a software for regulatory motif discovery using correlation with rank lists by Jacobsen2010. It uses a Binomial model to calculate the probability \( P_w(X\leq x) \) of word occurrence in a sequence where the specific word probability is derived from a Markov model on shuffled k'mers of the sequence to derive the background distribution. The over-representation scores (OR-scores) \(s_{w,i} = -\ln (P_{w,i}(X\leq x)) \) of word \( w \) in sequence \( i \) are sorted according to the ranking list. Finally, a running sum of the sorted OR-scores is used in connection with a variation of the Kolmogorov-Smirnov test to find correlated words. In case of a correlation between the ranking list and the OR-scores, the running sum function shows a directed variation from a random walk like curve. The background distributions of ranked sum curves are derived by permutation of the sequences. Cwords was run with standard parameters using word sizes of 6,\,7 and 8nt.
Cwords reports two motifs within the aligned top counting words which correlate with inaccessible sequences and two motifs correlating with accessible sequences.
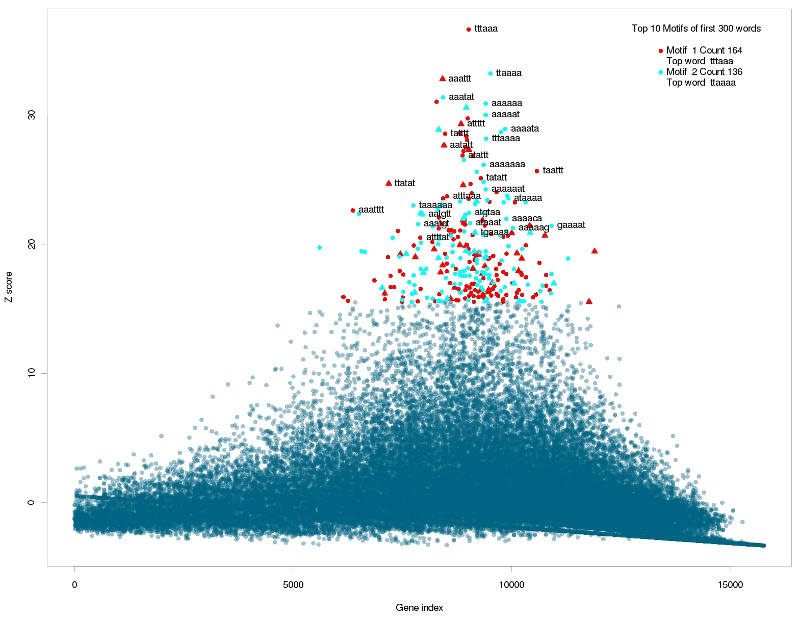
Correlated words overrepresented in accessible sequences.
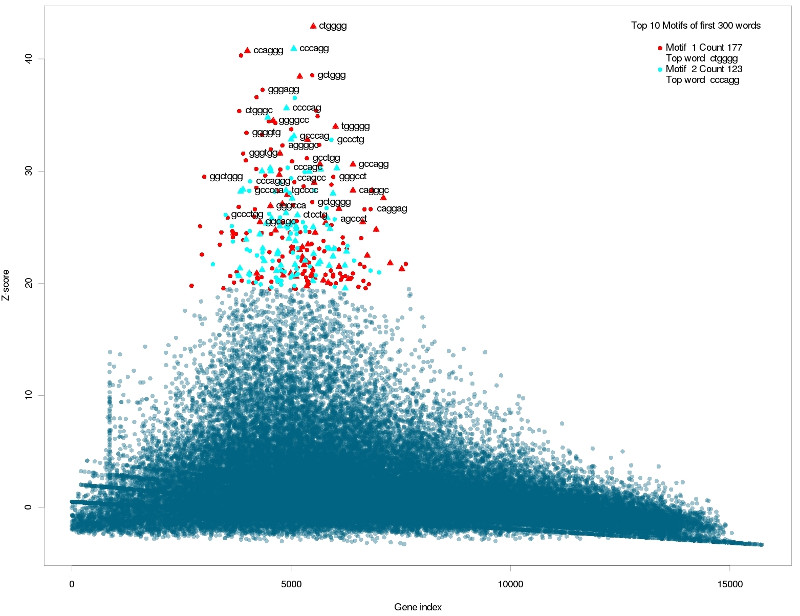
Correlated words overrepresented in inaccessible sequences.
They are GC rich words for inaccessible targets and AU rich words for more accessible sequences. Of interest in this matter is how well the global accessibility score \( GA(S_i) \) correlates with the energy needed to open the target structure \( \Delta G_{open} \). The Pearson correlation coefficient is 0.71 but the data shows a strong non-linearity. A linear regression shows that the residuals are not normally distributed (Shapiro-Wilk test: \( p\ll 0.01 \) ) and are not independent and show great auto-correlation. To evaluate the differences, the target sequences were ranked by the \( \Delta G_{open} \) score and the analysis was re-run using cWords with standard parameters. The only difference is that the over-represented words correlate less strong with the \( \Delta G_{open} \) score than with the global accessibility score \( GA \). But the words actually showing correlation are again CG rich for sequences with energy-demanding sequences (results not shown).
Focus on targets within hairpins
As reported by Kedde et al., the experimentally approved structural switch includes a hairpin structure. Therefore, it is assumed that hairpins play an important role within a potential structural switch and the next step was to move focus to miRNA targets within hairpin structures. The mfe structure was predicted of all 3'UTRs targeted by one of the 54979 predicted miRNA target sites. Regular expressions were used to search bracket representation for miRNA sites targeting hairpin structures within a sliding window of 37nt. A valid hairpin was defined as:
- the miRNA seed site is within the valid hairpin and has maximal one unpaired base
- the miRNA seed site can be followed by up to 5 paired bases
- the hairpin needs to have a loop of minimum 1 unpaired nucleotide
- the opposite sequence of the stem is allowed up to two unpaired bases
The analysis leads to a total of 2613 miRNA seeds within valid hairpins. Having done a Fisher test as for miRNA family miR-221/222 in the following table gave a list of most unbalanced microRNA families where 21 miRNA families have p-values smaller than 0.05. It needs to be mentioned that the p-values are not corrected for multiple testing. The microRNA showing the greatest enrichment within hairpins is miR-410.
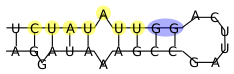
Example of a secondary structure of a valid hairpin. The miRNA seed site (yellow) needs to be within the valid hairpin and is allowed one unpaired base. The seed site can be followed by up to 5 paired bases (blue) followed by the loop of minimal length of 1nt. The oposite sequence of the stem is allowed to have up to two unpaired bases.
| miR221/222 | other miRNA | |
| hairpin | 24 | 2613 - 24 = 2589 |
| other | 295 | 52366 - 295 = 52071 |
Fisher test of miRNA family 221/222 targeting hairpins gives a p-value of \( \approx \) 0.025
Interrelation of targeted hairpins and over-represented words.
The further analysis concentrated on the data-set of 2613 seed sites within valid hairpins. This set was further reduced to a maximal independent set of 2235 sequences. The following figure shows the distribution of \( \Delta G_{open} \) of the subset of valid hairpins compared to the distribution of all seed sites. The Wilcoxon test shows that both distributions differ significantly ( \( p<2e-16 \) ).
The main fraction of hairpin structures scores in a range of -12 to -2. To find over-represented words within the hairpins which correlate with the energy score \( \Delta G_{open} \) a sequence set was extracted of 30nt long sequences not including the microRNA target site following the direction of the hairpin. The 300 most correlated words reported by cWords were aligned and visualised using Weblogo [Crooks2004]. Within the top 10 motifs is also one AT rich motif with an average z-score of \( \tilde{z}=3.01 \) standing out of the otherwise GC rich motifs.
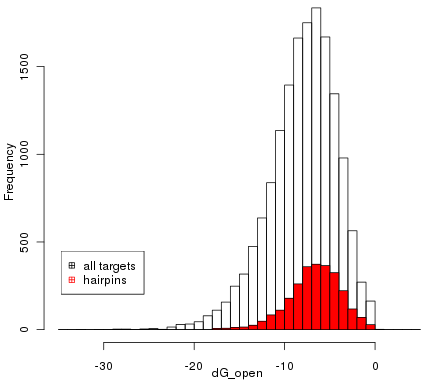
Distribution of \( \Delta G_{open} \) of all target sites compared to the subset of valid hairpins (red).
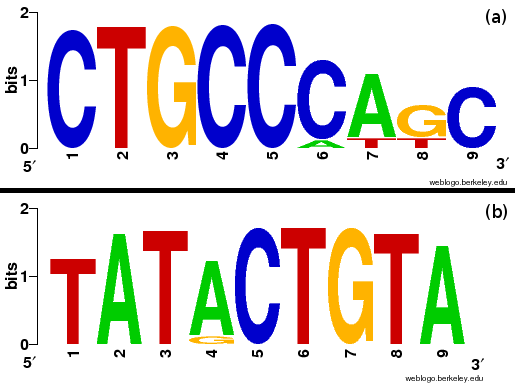
Motifs are based on aligned over-represented words as reported by cWords. Shown are only results of words which show positive correlation with the energy score \( \Delta G_{open} \). Analysis and clustering was done using cWords on a maximal independent set of 2235 sequences forming valid hairpin structures.
microRNA related motifs within hairpins
Next, I focused on subsets of hairpins by microRNA family showing great imbalance according to the top of table S1. The identification of over-represented motifs was done using self-organising maps and the software SOMBRERO v.1.1 [Mahony2005a,Mahony2005]. Further see Self Organising Maps.
The self organising map consists of a grid of MxN nodes where each node has the coordinates \( s=(s_1,s_2) \) and is described by a certain position weight matrix (PWM) of a motif of length \( l \). The PWM represents a feature of the input sequences where \( p(b,i) \) is the probability of having nucleotide \( b \) at position \( i \) compared to the background probability \( p(b) \). \[ PWM(b,i) = \log_2\frac{p(b,i)}{p(b)} \]
After the training, all the PWMs span over the entire feature space of the input sequences due to unsupervised learning. The PWMs of the SOM grid are first initialised using random values. SOMBRERO uses a more structured approach for initialisation but the random approach does not affect the motif-finding accuracy and the main idea of the SOM [Mahony2005]. During the training the input sequences are segmented into overlapping sub-sequences of length \( l\). Batches of these sequences are associated in a incremental fashion to the closest node on the grid by maximum likelihood, i.e. the highest scoring PWM. A motif can be easily scored by summing the position specific values of the weight matrix. A position count matrix (PCM) keeps track of the composition of associated sequences and is used to update the PWM of the node after each round of training. Additionally, neighbourhood nodes are also updated using a distance specific weight \( \Phi |s-s'| \) (in case of SOMBRERO the Gaussian neighbourhood function is used: \[ \Phi(|s-s'|) = \exp(-[(s_1-s'_1)^2+(s_2-s'_2)^2]/\gamma) \]
The influence of neighbour nodes is defined by the parameter \( \gamma \) which decreases during the cycles and reduces the influence throughout the training. After repeating the training cycle until a convergence is met, the grid spans over the feature space of all input-sequences and each sequence is associated with its closest node. To find over-represented motifs a random set of input sequences is used to derive the expected number \( E(n_i) \) of sequences associating with a certain node \( i \) of the SOM. Using multiple random sets also gives the standard deviation \( \sigma_i \) at each node. These random sets derive from a Markov chain based model.
Finally, a z-score is used to rank the motifs. \[ z = \frac{n_i - E(n_i)}{\sigma_i} \] As a background, a 3th order Markov chain was trained on a set of sequences obtained by extracting \( \pm \) 250nt around each target site of a certain miRNA family. SOMBRERO was used to find over-represented motifs searching for words of length 6 and 8. Results of interest are shown in the supplementary material in figure S2.
Interrelation of Pumilio and hairpins
Pumilio became a subject in recent research related to microRNA function [Triboulet2010,Kedde2010,Leibovich2010]. The aim of the further analysis is to characterise a possible interrelation of PUM2 binding sites and their cooperative behaviour in relation to hairpin structures. The PUM2 binding profile was obtained from Hafner2010 and converted into a PWM. All 3'UTR sequences were scored using the PUM2 PWM and matches with a score greater than 10 were reported which led to a total of 11,888 potential PUM2 binding sites. To classify the amount of PUM2 binding sites, all 3'UTR sequences were taken which have a potential binding site for PUM2. This led to a sequence of length 12,428,774 where a 5th order Markov chain was trained on as a background model.
The Poison distribution was used to test the Null-hypothesis that the occurrence of PUM2 binding sites is just by chance due to the composition of the sequences (no correction for multiple testing). The results show PUM2 sites which are imbalanced within hairpin structures in general.
PUM2 sites were linked with microRNA targets within hairpins and PUM2 sites within annotated hairpins plus/minus 30nt were reported. The following table shows the 9 microRNA families which have an imbalanced amount of PUM2 binding sites within microRNA targeted hairpins based on the Fisher-test (uncorrected p-values). Finally, the Fisher-test was used to answer the Null-hypothesis that the distribution of PUM2 motifs within hairpins is independent of the annotation by microRNA target sites. The following table shows the distribution of PUM2 motifs and the Fisher test gives a p-value of 9.05E-3 which means that there are significant more PUM2 motifs within hairpin structures if they are annotated microRNA targets.

| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |
| U | 1.89 | -3.1 | 1.89 | -3.46 | 0.59 | -3.36 | 1.74 | 0.05 |
| G | -3.39 | 1.75 | -3.39 | -3.46 | -1.41 | -3.36 | -3.04 | -2.58 |
| A | -3.39 | -1.43 | -3.39 | 1.9 | 0.8 | 1.89 | -1.28 | 1.4 |
| C | -3.39 | -3.1 | -3.39 | -3.46 | -1.41 | -3.36 | -3.04 | -2.58 |
The binding profile of PUM2 derived from PAR-CLIP by Hafner et al. and the translated PWM. A pseudo-count of 0.05 bit was applied before \( \log_2\frac{R_{i,j}}{\mu(R_j)} \) transformation. ( \( R_{i,j} \) is the information content of nucleotide \( i \in \) A,U,G,C at position \( j \); \( \mu(R_j) \) is the average informational content of nucleotides at position \( j \) ( \(\frac{\sum_iR_{i,j}}{4} \) )
| motif | other struct | hairpins | p-value |
|---|---|---|---|
| UGUAAAAA | 1121 | 399 | 5.89E-3 |
| UGUAAAUU | 986 | 360 | 3.18E-2 |
| UGUAUAUA | 1269 | 664 | 1.82E-7 |
Imbalanced PUM2 binding motifs within valid hairpin structures. This includes all predicted hairpins regardless of being targeted by microRNAs. The p-value derives from the Fisher-test. The top of the table presents motifs over-represented in hairpins and the bottom of the table under-represented motifs.
| miR annotated | other struct | hairpins |
|---|---|---|
| FALSE | 6210 | 2477 |
| TRUE | 3322 | 1469 |
The distribution of PUM2 motifs in relation to microRNA target site annotation and valid hairpin structures. Fisher-test gives a p-value of 9.05E-3.
| miR | hairpins | Pum2 | p-value |
|---|---|---|---|
| miR-300 | 70 | 38 | 1.01E-11 |
| miR-410 | 59 | 24 | 4.91E-6 |
| miR-181 | 63 | 19 | 1.80E-3 |
| miR-340/340-5p | 75 | 19 | 7.07E-3 |
| miR-144 | 46 | 13 | 1.14E-2 |
| miR-9 | 40 | 0 | 1.86E-2 |
| miR-329/362-3p | 16 | 6 | 2.81E-2 |
| miR-125/351 | 36 | 0 | 2.82E-2 |
| miR-124/506 | 49 | 1 | 3.79E-2 |
Annotated hairpins with potential PUM2 binding sites. The p-value derives from the Fisher-test.
Gene Ontology
The Gene Ontology database [Consortium2000] is an effort to define a standardised vocabulary to annotate genes and gene products. A great fraction of genes and gene products are equal or similar between all eukaryotes sharing the same core functions. This knowledge about the biological function of shared gene products can therefore be transferred in most cases. The GO database has three domains: biological processes, molecular functions and cellular components. The Ontology is organised as a directed acyclic graph which means that vertices are connected through directed edges without forming directed cycles. The Gorilla web service was used [Eden2009] to determine whether microRNA targets within hairpins show any enrichment compared to microRNA targets outside of hairpins. An enrichment of a specific GO term is calculated by: \[ enrichment = \frac{b/n}{B/N} \] where \( b,B \) are the amount of genes associated with a specific GO term and \( n,N \) are the total number of genes associated with any GO term. Capitalised letters represent the total amount of genes in target and background set and small letters stand for target set related measures.
The probability of the enrichment derives from a Fisher's test. The hypogeometric distribution is a version of the binomial distribution without replacement where the hypogeometric tail describes the probability of having \( b \) or more successes (specific GO annotations) in a sequence of \( n \) draws out of a population of size \( N \) with \( B \) of this specific GO term: \[ P(X\geq b) = \sum_{i=b}^{\min (n,B)} \frac{\binom{n}{i}\binom{N-n}{B-i}}{\binom{N}{B}} \] As a remark, this p-value is not corrected for the multiple testing of GO-terms. GO-terms with a p-value smaller than 1E-3 were reported. Further, the REViGO web service [Supek2010] was used to cluster GO terms based on similarity measures [Schlicker2007]. All reported GO terms were used with their p-values and a distance threshold of 0.7. The first target set are 1784 gene IDs with microRNA seeds within valid hairpin structures where 1667 are annotated with a GO term. The background set consists of 7383 gene IDs with microRNA seeds in other kind of structures where 6894 are annotated with a GO term.
MicroRNA seeds within hairpin structures show an enrichment of genes related to biological processes taking part in
- Notch signaling pathway
- pattern specification processes
- developmental processes
- biological regulation
- chromatin modification
They show an enrichment in their molecular functions of
- sequence specific DNA binding RNA polymerase~II transcription factor activity
- nucleic acid binding
- zinc ion binding
- nucleic acid binding transcription factor activity
and are related to cellular components such as the
- PcG protein complex



The second target set was the sub-set of 273 gene IDs of microRNA seeds within hairpin structures having a predicted PUM2 binding site in the neighbourhood of 30nt of the hairpin were 251 are GO annotated. The background set consists of 8894 gene IDs of microRNA seeds not being part of the target set were 8561 are GO annotated. The PUM2 related hairpin targets show enrichment for the biological process of
- regulation of mesenchymal cell proliferation
They are enriched in their molecular functions of
- nucleic acid binding
- zinc ion binding
- molecular function
- retinoic acid 4-hydroxylase activity
And are related to the cellular component:
- PcG protein complex



Discussion
The aim of this thesis was at first to build a model of a RBP introduced structural switch mediating microRNA accessibility of 3'UTR sequences. It turned out that there was only one reported example [Kedde2010] and it became obvious that the focus of the analysis had to change to a more general sequence analysis. The publication of the structural switch proposes three interesting domains: microRNA regulation, structural composition of target sites and interaction of RBPs. The new aim became to characterise the interplay of this three subjects.
The first interesting article showing up during the literature research was by Leibovich et al. which introduces the global accessibility score as a structural approach and describes a cooperative activity of PUM1 and miR-410.
The intuitive idea was that highly conserved but inaccessible microRNA targets should show some cooperative behaviour with RBPs to overcome the barrier of their inaccessible targets. The global accessibility score \( GA \) was meant to be a suitable measure for this approach, incorporating both: the overall composition of the sequence and the necessary free-energy difference to open the seed site.
CWords was used to find over-represented words which correlate with the ranking of the sequences by their global accessibility score. The method is based on an imbalance of words within a ranked list where the test statistic is based on a empirical background distribution. This makes it easily adoptable to ranking lists based on any kind of scoring.
As there were almost no information about the expected results this analysis was looking for, it was necessary to define some criteria not to drown within the flood of words and motifs. The decision was to focus on AU rich motifs which would stand out of the GC rich background of the highly inaccessible target sequences. This decision also agrees with the Pumilio binding site reported in the two articles the thesis started with Kedde2010,Leibovich2010.
Unfortunately, Cwords only shows a correlation of GC rich sequences with highly dense and inaccessible structures and that open structures are AU rich. This result was expected due to the 3 hydrogen bonds of GC base pairs. An AU rich motif as reported in Jacobsen2010 did not show any significant correlation with inaccessible sequences.
The further analysis showed that the relationship between global accessibility score \( (GA) \) and \( \Delta G_{open} \) is non-linear. The interpretation of this results is that the signal of the 3 hydrogen bonds of GC rich words is too strong and overrules the probably weaker AU rich signal correlating with the \( \Delta G_{open} \) part. The global accessibility score is given by: \[ GA(S_i) = 2\Delta G_{all}(S_i) - \Delta G_{masked}(S_i) \] which shows the dominant character of the general sequence composition. The sequence composition is of course somehow related to structural properties of a sequence and therefore to their accessibility. But the concern is that the \( GA \) score is too dependent on the GC content of the sequence.
To test this hypothesis the sequences were ranked according to their \( \Delta G_{open} \) value. The only difference in the results was that the GC rich words have lower z-scores and show weaker correlation than before. The conclusion is that the structural features - which are probably not restricted to distinct sequence motifs - are overcome by the GC prone energy scores.
As the approach did not give anything of interest the three domains need to be narrowed down and the further analysis focused on hairpin structures. This decision was motivated by the one experimental approved example of a structural switch including a hairpin structure. If the hairpin structure is actually a necessary feature of the structural switch, it explains why the correlation of the global accessibility score \( GA \) was unsuccessful. Focusing on over-represented words showing greatest energy barrier for opening the target sites do not include hairpin structures.
The definition of a valid hairpin is strongly orientated at the hairpin reported by Kedde et al. It turns out to be very strict but as there is no further data to base any evaluation of hairpin criteria on, any decision seems as good as the other.
The analysis leads to 2613 microRNA seed sites within hairpin structures. The mfe structure predicted by RNAfold does not necessarily represent the folding in vivo. As a limitation of the thermodynamic models, the prediction accuracy decreases with the sequences length. The prediction was made on full 3'UTR sequences which varied highly in length within a range of several 10k. This obviously means that not all of the predicted hairpins actually fold in vivo and that there are probably some missing.
After the data-set was narrowed down to seed sites within hairpins, cWords reports one AU rich motif of 9 over-represented words which show correlation with the \( \Delta G_{open} \) score.
The Results show that 21 microRNA families present strong imbalance of target sites within hairpin structures. There is certainly a risk of false positives as the Fisher tests were not corrected for multiple testing. It needs to be discussed whether the significance criteria of p-value \( > 0.05 \) is sufficient. In a next step the data-set was further split into subsets by microRNA family. The motivation was that regulatory motifs are maybe microRNA specific.
The method of choice was SOMBRERO which is based on a SOM. The SOM seams to be eligible because of several reasons: On the one hand, the SOM is a semantic mapping which is used to reduce the dimensionality of words to motifs. This is motivated by the fact that RBPs have less specific seed sites which makes simple word analysis undesirable. On the other hand, the SOM is an unsupervised learning method which makes it very flexible regarding sequence requirements. The map adopts to the complexity and distribution of sequence features with more nodes of the map pointing towards parts of the input space of higher complexity. Also, there is only one parameter to adopt SOMBRERO to the characteristics of the specific input sequences which is the amount of nodes in the map. Finally, the trained map can be used to calculate a significance of the learned sequence motifs.
The results show over-represented motifs selected for PUM2 which SOMBRERO reports within the top 3 scoring motifs. The results show strong evidence for a Pumilio like motif. Also, cWords reports parts of a Pumilio binding site at position 6-9 of the AU rich motif. The analysis gave enough evidence to limit the RBP to Pumilio and to examine the role of Pumilio in relation to microRNA seed sites within hairpins.
The scoring of all 3'UTR sequences shows that the highest scoring PUM2 binding motif is under-represented compared to the background probability which gives evidence to evolutionary pressure on PUM2 motifs because of their role in 3'UTR targeting.
Compared to the amount of PUM2 motifs found, it shows that 2 motifs are especially enriched and 1 under-represented within hairpin regions. Interestingly, it is the highest scoring motif UGUAUAUA again which is also particularly under-represented in hairpins.
Furthermore, the microRNA families which show significant imbalance of PUM2 binding sites within hairpins. The top PUM2 enriched hairpins are annotated by microRNA families which also show over-represented targets within hairpins in general, namely: miR-410, miR-300 and miR-340. The target site of miR-300 (CUUGUAUA) shows the beginning of a PUM2 binding site. The seed sites of the microRNA were masked but it could be that miR-300 target sites occur regularly in pairs. This means that the miR-300 seed site increases chances of getting a PUM2 binding site as it provides already 6 out of 8 nucleotides. This would influence their background probability and probably mean that PUM2 binding sites are not as over-represented as assumed. It needs to be verified whether PUM2 sites within miR-300 targeted hairpins are also miR-300 targets themselves. However, it does not change the fact that PUM2 binding sites are imbalanced towards hairpin structures as reported by the Fisher test.
A final Fisher test shows that significantly (p=9.05E-3) more PUM2 binding sites are within hairpins if they are also annotated by microRNA seed sites.
Contrary, there are two microRNA families showing preference for hairpins but are underrepresented with PUM2 targets, namely: miR-141/200a and miR-124/506. This two microRNA families are good candidates for other RBPs showing cooperative behaviour with microRNA targets within hairpins.
The results of the gene ontology analysis show that all enriched GO terms in all three domains of the sub-set of PUM2 targeted hairpins with microRNA seeds are also a sub-set of the enriched GO terms of all microRNA targeted hairpins. Especially, hairpins with PUM2 binding sites show an enrichment in genes related to the regulation of mesenchymal cell proliferation and are related to the PcG protein complex. The PcG protein complex is a chromatin-associated multiprotein complex which is know to maintain long-term gene repression in Drosophila to preserve body patterning through development by influencing chromatin structure [Francis2004]. Mesenchymal cells are multipotent stem cells which can differentiate into fat cells, bone cells and cartilage cells.
It is presumed that there are other RBPs associated with microRNAs within hairpins. The GO terms suggest that the genes not targeted by PUM2 are related to the Notch signaling pathway and encode transcription factors.
Summary
There are certain miR families which show enriched seed sides within hairpin structures. The genes forming this hairpins show a relation to the PcG protein complex which is important for long-term gene repression during development of Drosophila. Additionally, some of the hairpins with microRNA seed sides show binding sites of PUM2. PUM2 seems to have a specific cooperative behaviour towards hairpin structures with microRNA seed sides. Particularly, miR-410, miR-300, and miR340 are potentially main co-partners of PUM2. The genes regulated by this microRNA targeted PUM2 hairpins are involved in the regulation of mesenchymal cell proliferation through the PcG protein complex.
Whether the cooperative behaviour of PUM2 with this microRNA seeds within hairpins incorporates a structural switch cannot be assessed by this analysis. But it is a likely assumption as it is experimental shown for one example where this analysis was based on. Additionally, cWords reports a potential PUM2 binding motif which is particularly enriched within hairpins showing greatest inaccessibility based on the \( \Delta G_{open} \) energy score.
Supplementary material
S1: Distribution of miRNA targets within hairpins with p-value<0.1 . The p-value derives from a Fisher test and reflects the imbalance between targets within and outside of hairpins. The p-value is not correct for multiple testing. The column all sites shows the total count of target sites, hairpins show the fraction of sites within valid hairpins and p-value the p-value of the Fisher test.
| miRNA family | all sites | hairpins | p-value |
|---|---|---|---|
| miR-410 | 475 | 59 | 2.28E-11 |
| miR-410 | 475 | 59 | 2.28E-11 |
| miR-300 | 715 | 70 | 1.46E-08 |
| miR-374/374ab | 499 | 42 | 4.19E-04 |
| miR-592/599 | 146 | 0 | 1.41E-03 |
| miR-494 | 375 | 32 | 1.41E-03 |
| miR-340/340-5p | 1094 | 75 | 1.94E-03 |
| miR-140/140-5p/876-3p | 253 | 3 | 4.24E-03 |
| let-7/98 | 907 | 26 | 5.70E-03 |
| miR-124/506 | 1479 | 49 | 7.58E-03 |
| miR-1/206 | 639 | 17 | 1.11E-02 |
| miR-290-5p/292-5p/371-5p | 208 | 18 | 1.34E-02 |
| miR-133 | 530 | 14 | 1.80E-02 |
| miR-141/200a | 580 | 40 | 1.83E-02 |
| miR-96/1271 | 866 | 27 | 1.95E-02 |
| miR-137 | 949 | 30 | 2.05E-02 |
| miR-128 | 865 | 27 | 2.36E-02 |
| miR-221/222 | 319 | 24 | 2.46E-02 |
| miR-370 | 246 | 19 | 3.47E-02 |
| miR-182 | 927 | 31 | 4.26E-02 |
| miR-194 | 269 | 20 | 4.39E-02 |
| miR-34a/34b-5p/34c/34c-5p/449/449abc/699 | 515 | 15 | 4.76E-02 |
| miR-217 | 251 | 19 | 5.07E-02 |
| miR-504 | 148 | 2 | 5.07E-02 |
| miR-758 | 149 | 2 | 5.10E-02 |
| miR-145 | 589 | 18 | 5.11E-02 |
| miR-144 | 724 | 46 | 5.22E-02 |
| miR-181 | 1039 | 63 | 5.50E-02 |
| miR-139-5p | 259 | 19 | 5.63E-02 |
| miR-382 | 130 | 11 | 5.92E-02 |
| miR-216/216a | 163 | 13 | 6.27E-02 |
| miR-149 | 277 | 20 | 6.38E-02 |
| miR-543 | 536 | 35 | 6.51E-02 |
| miR-30a/30a-5p/30b/30b-5p/30cde/384-5p | 1241 | 73 | 6.79E-02 |
| miR-590/590-3p | 907 | 32 | 8.32E-02 |
| miR-136 | 153 | 12 | 8.33E-02 |
| miR-135 | 546 | 17 | 8.44E-02 |
| miR-18ab | 197 | 4 | 8.99E-02 |
| miR-431 | 99 | 1 | 9.38E-02 |
| miR-22 | 343 | 23 | 9.64E-02 |
S2: over-represented motifs in hairpin structures of miR family specific subsets





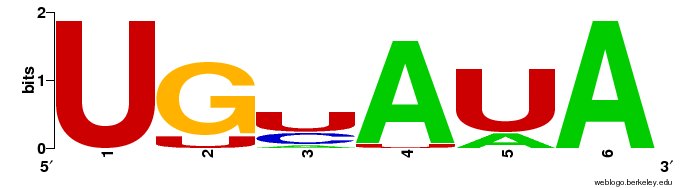

S3: Potential PUM2 binding motifs and their occurrence within 3'UTR sequences. The background probability \( p(bg) \) derives from a 5th order Markov model trained on a background sequence of all 3'UTR with potential PUM2 binding sites (length: 12,428,773). The reported p-value derives from the Poison test.
| score | 10.24 | 13.26 | 11.91 | 11.05 | 11.05 | 10.03 | 13.05 | 11.7 | 10.17 |
| word | UGUAAAAA | UGUAAAUA | UGUAAAUU | UGUACAUA | UGUAGAUA | UGUAUAAA | UGUAUAUA | UGUAUAUU | UAUAAAUA |
| occurrence | 1483 | 2157 | 1317 | 1205 | 520 | 1287 | 1904 | 1649 | 1688 |
| \( p(bg) \) | 3.08E-4 | 1.06E-4 | 9.32E-5 | 4.95E-5 | 2.32E-5 | 8.49E-5 | 1.15E-4 | 9.84E-5 | 8.66E-5 |
| p-value | 2E-16 | 2E-16 | 4.85E-6 | 2E-16 | 2E-16 | 4E-12 | 2E-16 | 2E-16 | 2E-16 |